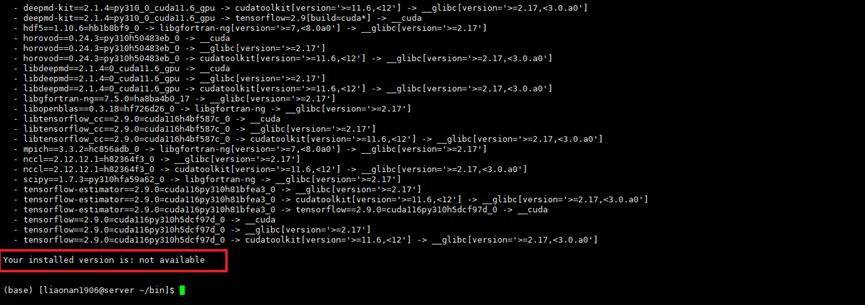
和收敛有关的参数主要有能量的CUTOFF,SCF的次数,SMEAR,以及K点选取等,可以尝试一下。 SCF最大仍不收敛 一般来说,MS默认的SCF次数100对于小系统是够用的,但是,原子数一多,就不一定了。 a) 首先结构的合理性,如果自建的结构偏离最低能量太大(或失配度太大),可能会难以收敛。 b) 取消一切对称性,充分驰豫晶格。 c) 有些结构本身就是亚稳态,scf的各项收敛指标如果设的太高,对于亚稳态就可能达不到如此的精度。 d) 改变收敛的条件,降低精度(能量的截断值)。 e) 增加循环次数。 f) 改变赝势。 优化不收敛 增加设Max.Interations的大小 interations 是定义积分精度的,相当于gaussian里的int选项;根据gauss的经验,对重原子如果不用细的积分网格,结果就不准确,特别是频率计算,可能会把正的频率算成负的频率。提高 interations的确可以提高精度,特别是对于过渡态和频率计算。 总能和文献不一致,而且多种方法都不同 很正常,只要相对值近似就可以。 在倒空间进行电子结构计算的方法中,都存在一个能量零点的取法的问题,不同的程序,取的不同。根本原因是V(G)在G=0是发散的(可以去R.T. Martin的电子结构那本书)。 castep是利用周期性,在倒空间进行电子结构计算的.我没有记错的话,Dmol3是在实空间来进行电子结构计算的.能量零点的取法就跟不同前者了。 因此在进行电子结构计算中,不要比较任何绝对能量值的大小,这样是毫无意义的,即使是同一个程序中也是这样,更不用说是两个不同的程序。 能量的相对值才具有物理意义。