二维材料无法收敛的问题 NELMDL的设置
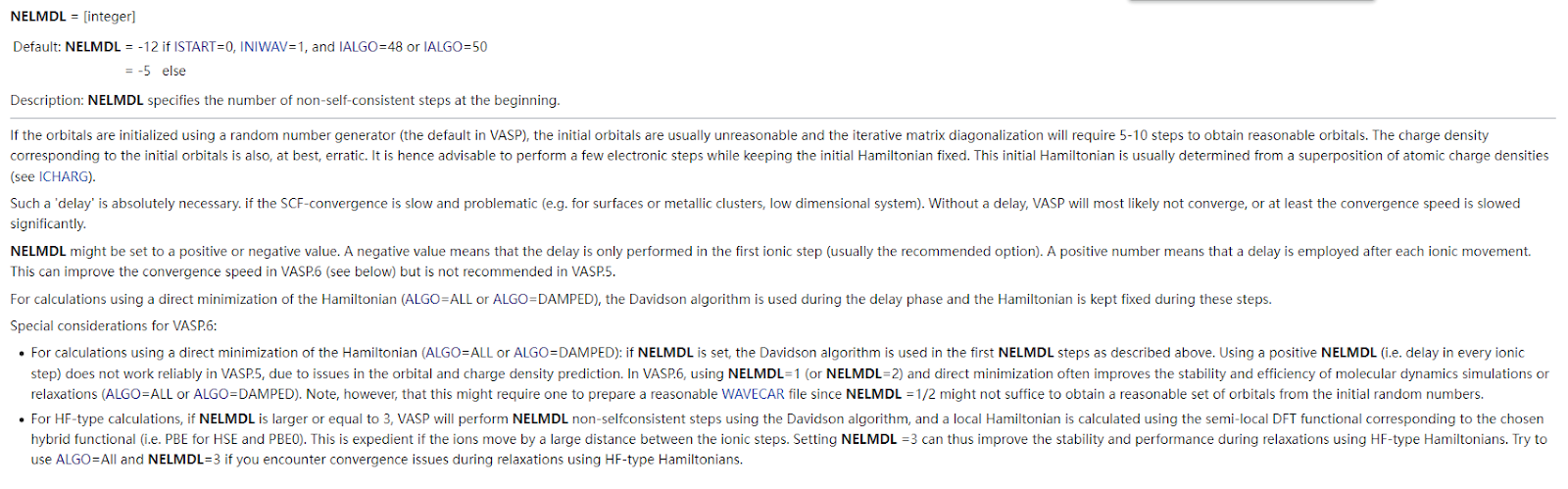
在计算二维材料单点能计算的时候发现120个电子步后仍无法手链,一番请教老师过后,发现可以尝试修改NELMDL这个参数 有关NELMDL参数的解释 一开始尝试NELMDL=-12和-18,并没帮助收敛(其中-18在108个电子步才收敛),这里说在vasp6以上版本使用ALGO=all和NELMDL=1会帮助很快收敛,然后我尝试修改这两个参数,发现确实可以帮助收敛,40个电子步就可以快速收敛了 (而且只花费了2600s,不到一小时) ,但是我仔细查阅ALGO=all这个参数发现 这个算法似乎对于绝缘体比较好用,但是不知道能否用于半导体,持着谨慎的态度又讲ALGO改回了Normal,发现也能够收敛,不过需要50步左右的电子步,所以我们还是选择这个参数会比较保险一点。